Il Linfoma: in questo articolo capiremo cos’è il linfoma, quali sono i sintomi e le cause del suo sviluppo, la diagnosi con le tipologie diverse di linfoma e i trattamenti da preferire in questi casi. Si tratta di un vero e proprio tumore collegato a problematiche relative al sistema immunitario: ma vediamo, nello specifico, tutto ciò che c’è da sapere sui linfomi.
Linfoma: Cos’è?
Il termine linfoma indica una serie di tumori che colpiscono il tessuto linfoide, ovvero i linfociti T, i linfociti B ed i linfociti Natural Killer (NK). I linfomi sono quindi un insieme di neoplasie che colpiscono il sistema immunitario (tumori solidi), ma non il sangue. I linfomi non sono necessariamente maligni, ma questo termine viene ormai utilizzato solo per definire i tumori che presentano caratteristiche di malignità.
I linfomi sono stati descritti a partire dal 1832 dal medico Thomas Hodgkin, che diede il nome ad una classificazione che è stata tuttavia successivamente espansa ed integrata dall’OMS per comprendere anche tumori linfoidi dalle caratteristiche istologiche particolari. Ad oggi, i linfomi sono tra i tumori maligni più diffusi al mondo (5% di tutte le diagnosi).
[boxinformativo title=”I linfomi rappresentano la quinta causa di morte per cancro nel mondo.”][/boxinformativo]
Linfoma: Sintomi
I linfomi causano sintomi aspecifici, difficilmente collegabili a questa malattia in assenza di analisi di laboratorio. I sintomi iniziali del linfoma sono:
- abbondante sudorazione notturna che causa il risveglio
- forte ed inspiegabile perdita di peso in poco tempo
- febbre altalenante anche alta
- anemia
- astenia
- edema
- ipercalcemia
- epatosplenomegalia
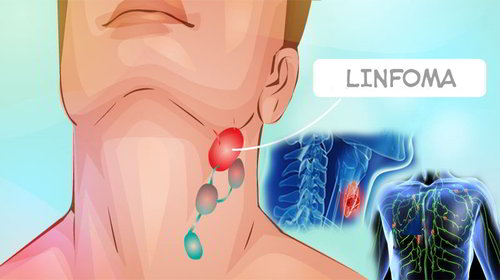
- ingrossamento linfonodale senza dolore
- prurito
- masse e tumefazioni nei siti corporei colpiti
- piastrinopenia

[boxinformativo title=”I linfomi non sono contagiosi: stare a contatto con un malato di linfoma non è pericoloso.”][/boxinformativo]
Ci sono altri sintomi più specifici che però appaiono a malattia avanzata: ascite, dispnea, dolore al petto, ittero, nausea, infezioni batteriche gravi, paraplegia, pancitopenia, trombocitosi, versamento (pleurico o pericardico), sindrome nefrosica, calcoli renali, proteinuria.
Linfomi: Cause
Ad oggi, l’eziologia dei linfomi è per la maggior parte dei casi (70%) sconosciuta. Sono state formulate molte ipotesi per spiegare l’intensa ed incontrollata attività di proliferazione delle cellule immunitarie: è noto, ad esempio, che fino al 20% dei linfomi può essere provocato da infezioni sia batteriche che virali. L’Helicobacter pylori, un batterio che risiede nello stomaco, può causare il linfoma follicolare primitivo dello stomaco; il virus di Epstein-Barr può provocare il linfoma di Burkitt; il virus dell’epatite C ed il virus HTLV-1 possono causare la leucemia dell’adulto a cellule T o il linfoma non Hodgkin.
Anche il calo di difese immunitarie può essere la causa dell’insorgenza di un linfoma, si calcola fino al 5% dei casi. L’immunodeficienza può essere primaria, ovvero geneticamente determinata, o secondaria ad altre malattie come ad esempio HIV, farmaci anti-rigetto post-trapianto, terapia di malattie autoimmuni (sindrome di Sjogren, artrite reumatoide, lupus, tiroidite di Hashimoto). Solamente una piccola parte dei linfomi, meno dell’1%, è associato a fattori ambientali (inquinamento, radiazioni, pestidici, sostanze chimiche).
Come nella maggioranza delle altre tipologie di tumore, anche per i linfomi la causa scatenante è la sovraespressione di un oncogene abbinata solitamente all’inattivazione/silenziamento di uno o più geni oncosoppressori. Gli oncogeni sono geni in genere deputati al controllo della proliferazione cellulare e sono per questo attentamente controllati da complessi meccanismi, in quanto la loro azione è determinante nella trasformazione della cellula normale in cellula cancerosa; molto spesso una traslocazione dell’oncogene ne causa anche l’attivazione, in quanto esso si sottrae al controllo di tali meccanismi. I geni oncosoppressori sono geni che al contrario reprimono la proliferazione cellulare e quindi sono sempre attivi; una mutazione nei suoi domini regolatori può però impedirne la funzione, permettendo alla cellula di replicare senza alcun controllo e provocare un tumore.
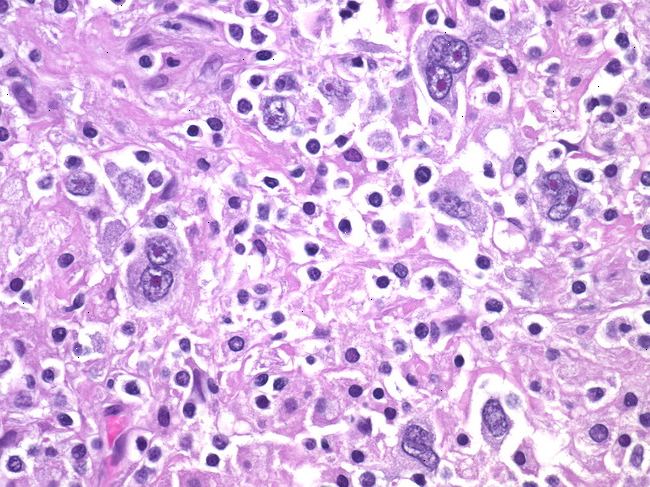
Diagnosi
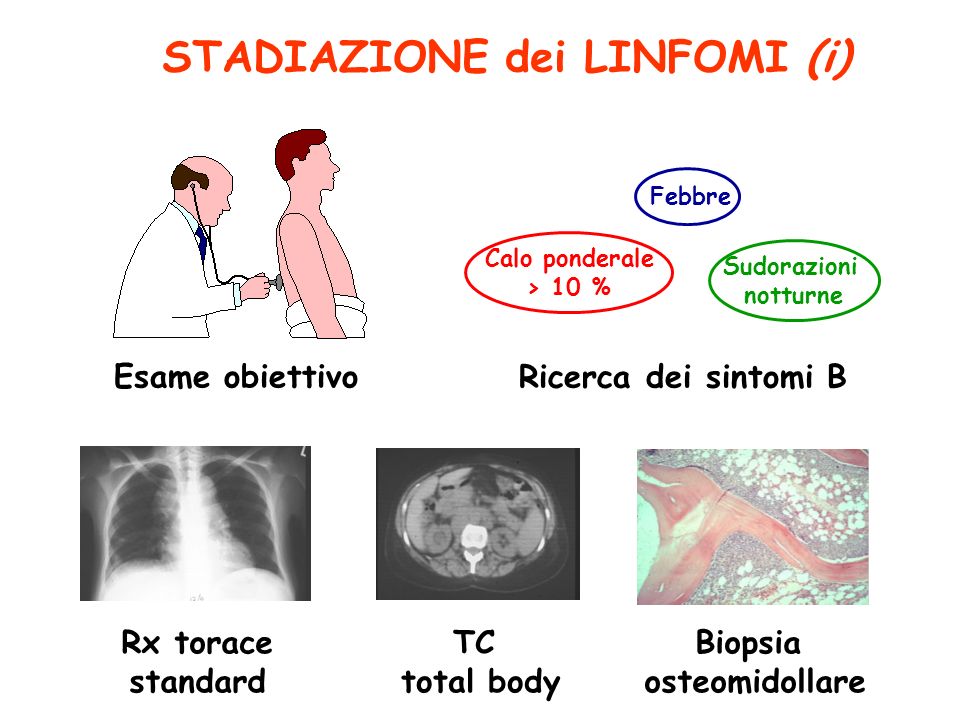
La possibilità di un linfoma dovrebbe essere presa in considerazione quando si ha un ingrossamento persistente e non doloroso di un linfonodo, insieme ad altri sintomi come sudorazione notturna e prurito. La diagnosi definitiva è sempre di laboratorio, tramite unabiopsia: un piccolo frammento del linfonodo viene asportato e sottoposto ad esame istologico, per analizzare l’architettura tissutale e le cellule e confermare la presenza di anomalie. L’esame istologico è inoltre fondamentale per classificare il linfoma in modo da scegliere la terapia più appropriata. Se l’ingrossamento riguarda un altro organo (tipicamente milza o fegato) la procedura è la stessa. La stadiazione della malattia è il passo successivo e serve a determinare l’estensione del linfoma. Si eseguono quindi ulteriori esami come il prelievo del midollo osseo, RX torace, TC total-body, risonanza magnetica, PET ed esami che riguardano i singoli organi per escluderne l’interessamento (gastroscopia, colonscopia, esame del liquor…). Sono inoltre necessari l’esame del sangue con elettroforesi delle proteine sieriche, l’immunofenotipo sul sangue periferico e midollare e analisi genetica delle cellule cancerose. Al termine della terapia, gli esami possono essere ripetuti per valutare l’efficacia del trattamento. (Vedi anche: Ematocrito)
Linfoma: Tipologie
La classificazione dei linfomi è molto ampia ed oggi la parola “linfoma”, come deciso dall’Organizzazione Mondiale della Sanità, indica più di 70 forme di tumori del tessuto linfoide, divise in 4 grandi sottogruppi.
Linfoma di Hodgkin (LH)
Neoplasia solida localizzata solitamente nei linfonodi assiali (cervicali, mediastinici). Tende a diffondersi tra linfonodi vicini tra loro ed è caratterizzato da tipiche cellule giganti (cellule di Reed-Stenberg) e dalla presenza di leucociti infiltrati nel tessuto tumorale. Esistono 3 forme di linfoma di Hodgkin: a prevalenza linfocitaria nodulare, classico (4 sotto-categorie a seconda della tipologia di cellule prevalenti) e inclassificabile (non rientra in nessuna delle altre classi). Non sono ancora state ritrovate cause certe per lo sviluppo del linfoma di Hodgkin, ma si sospetta un forte coinvolgimento del virus di Epstein-Barr in circa la metà dei casi. Pazienti HIV-positivi o con familiarità sono maggiormente a rischio. I sintomi della malattia sono linfoadenopatia con importanti ingrossamenti, febbre continua o ciclica (detta di Pel-Ebstein), prurito, sudorazione notturna, calo di peso, astenia, dolore diffuso, tosse (nella forma nodulare). Il linfoma di Hodgkin ha buona prognosi e può essere efficacemente trattato con cicli di chemioterapia e radioterapia.

Linfomi non Hodgkin (NHL)
Rappresentano circa il 90% di tutti i linfomi e colpiscono prevalentemente i linfonodi: sono quindi tumori solidi che occasionalmente possono interessare anche altri organi come il sistema gastro-enterico, la cute e le gonadi. A loro volta i linfomi non Hodgkin possono essere suddivisi in altre categorie che riflettono le differenze istologiche del tumore, dato che sul piano clinico la distinzione dei linfomi non Hodgkin da altre patologie (come le leucemie) è piuttosto difficoltosa. I linfomi non Hodgkin più comuni sono:
-
-
- Linfoma mantellare. Caratterizzato da una anomala iperproduzione di linfociti piccoli ed inefficaci, è molto simile al linfoma follicolare e ad alcune forme di leucemia linfocitica cronica. Si sviluppa nella zona mantellare del linfonodo – da cui il nome – ed il responsabile dell’insorgenza della patologia è nella stragrande maggioranza dei casi una anomalia cromosomica che attiva abnormemente il ciclo cellulare.
- Linfoma follicolare. E’ il linfoma non Hodgkin più frequente; solitamente generalizzato e a lento andamento, si sviluppa nella zona follicolare del linfonodo ed è caratterizzato da numerose cellule di piccole dimensioni (centrociti) e da poche cellule più grandi (centroblasti). Anche in questo caso la causa del linfoma è una mutazione cromosomica che immortalizza le cellule.
- Linfoma a grandi cellule B. Linfoma localizzato ed aggressivo causato da una mutazione che porta alla proliferazione cellulare. Può colpire sia i linfonodi che la cute, il sistema nervoso centrale e l’apparato gastro-intestinale e può derivare dal linfoma follicolare o dalla leucemia linfatica cronica.
- Linfoma di Burkitt. Tipico dell’età pediatrica e causato da una mutazione del gene c-myc. Può essere sporadico (20% dei casi, associato ad infezione da virus di Epstein Barr, coinvolte ileo e peritoneo), endemico (sempre causato da infezione da virus di Epstein-Barr, coinvolge mandibola e apparato urogenitale) o associato all’infezione da HIV (nel 25% dei casi co-infezione con virus di Epstein-Barr). E’ un linfoma aggressivo ma con ottime possibilità di terapia.
- Linfoma cutaneo. I linfomi non Hodgkin cutanei possono essere a cellule B o a cellule T, quest’ultimo chiamato anche micosi fungoide, che rappresenta la forma più comune di linfoma cutaneo; può evolvere nella sindrome di Sézary, che però secondo alcuni studiosi rappresenta un linfoma del tutto distinto. I linfomi cutanei sono difficili da riconoscere e si trattano topicamente se sono localizzati (chirurgia, radioterapia, fototerapia) oppure con chemioterapia se sono aggressivi o diffusi.
-
Mieloma multiplo
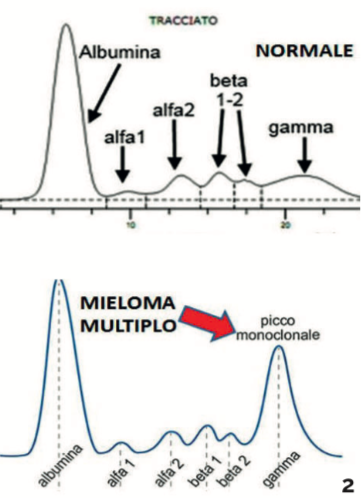
Neoplasia ematologica causata dall’anomala proliferazione delle plasmacellule, cioè linfociti B con la funzione di produrre anticorpi. Tipico dell’anziano, nel mieloma multiplo le plasmacellule cancerose sono colpite da mutazioni cromosomiche che attivano la proteina Rb ed il gene k-ras. I sintomi del mieloma multiplo sono dolori vertebrali, neuropatia, astenia, sudorazioni notturne, anemia, calo ponderale ed insufficienza renale. Analizzando l’elettroforesi delle proteine sieriche, tipico è l’innalzamento delle gamma-globuline prodotte dalle plasmacellule mutate (picco monoclonale).
Disordini immunoproliferativi
Le malattie immunoproliferative sono fortunatamente piuttosto rare e colpiscono prevalentemente le plasmacellule, le quali producono grandi quantità di frammenti anticorpali anomali ed in alcuni casi possono differenziarsi in altre classi di cellule B. Alcuni esempi sono:
-
-
- Linfoma maligno linfoplasmocitico. Tipologia di linfoma nel quale i linfociti tendono a differenziare in plasmacellule le quali producono massicciamente IgM che causano una sindrome dai iperviscosità (m acroglobulinemia di Waldenstrom) con rallentamento del flusso ematico nei vasi per il quale si rende necessaria la plasmaferesi (tecnica di centrifugazione del sangue per separarlo da elementi presenti in eccesso e quindi fluidificarlo). Fattori di rischio sono infezioni virali, familiarità, età avanzata; sintomi sono astenia, vertigini, cefalea, disturbi visivi. La terapia è solo palliativa e farmacologica.
- Amiloidosi primaria o β-fibrillosi . Patologia caratterizzata dalla deposizione nei tessuti di proteine insolubili (dette amiloidi per la tendenza a reagire con lo iodio, tipica dell’amiloide). Non ha cause note, ma è probabilmente associata a mieloma multiplo e linfoma benigno B; può colpire qualsiasi organo (tipicamente cute, cuore, lingua, nervi). I sintomi di conseguenza dipendono dagli organi colpiti: affaticamento e respiro corto nel caso del cuore, edema e gonfiore se sono colpiti i reni, aumento delle dimensioni della lingua, epatosplenomegalia. Le amiloidosi vengono classificate a seconda del precursore proteico che si accumula nei tessuti e nell’uomo ne esistono oltre 25 tipologie. Non esiste una terapia vera e propria; è possibile la chemioterapia e il trapianto dell’organo colpito in caso di pesante compromissione.
- Malattia delle catene pesanti. Patologia caratterizzata dalla produzione, da parte delle plasmacellule, di grandi quantità di frammenti di anticorpi definiti “catene pesanti”. Esistono diversi varianti di tale malattia e tutte le proteine prodotte in eccesso dalle plasmacellule sono geneticamente difettose; è probabile che la mutazione sia insorta durante la ricombinazione dei geni delle immunoglobuline, una tappa della maturazione linfocitaria. Tipicamente maligna e con una prognosi sfavorevole, la malattia delle catene pesanti può essere trattata con corticosteroidi e chemioterapici con risultati variabili.
-
Trattamenti
Le terapie dei linfomi dipendono dalla classificazione del linfoma, dalla stadiazione e dalle caratteristiche genetiche del linfoma.
-
Linfoma di Hodgkin
La terapia del linfoma di Hodgkin al primo stadio utilizza diversi chemioterapici, generalmente bleomicina, vinblastina, dacarbazina e doxorubicina. La chemioterapia è seguita da radioterapia nella zona colpita dal tumore. Se il linfoma è in stadio avanzato, la chemioterapia è la stessa ma con un maggiore numero di cicli. Dopo due cicli di chemioterapia si esegue una PET per verificare che il linfoma sia sensibile ai farmaci: se la PET è negativa, significa che le cellule neoplastiche sono sensibili ai farmaci e quindi è molto probabile la guarigione. Se il linfoma è resistente o ricompare dopo il trattamento (recidiva) si esegue una chemioterapia di seconda linea con farmaci diversi (gemcitabina, vinorelbina e ifosfamide). L’ultima frontiera terapeutica è rappresentata dal trapianto di midollo con cellule del paziente (autotrapianto) o da donatore, oppure dai nuovi anticorpi monoclonali (brentuximab) che ricoscono selettivamente le cellule tumorali, uccidendole.
-
Linfomi non Hodgkin
La terapia dipende dall’aggressività del linfoma e dalla presenza di sintomi:
-
Linfomi indolenti
Asintomatici o scarsamente sintomatici, possono non richiedere terapia ed essere tenuti sotto controllo per molto tempo con esami periodici; in casi selezionati può essere impiegata la radioterapia nell’area colpita, oppure l’associazione di anticorpi monoclonali (rituximab) e chemioterapia con ciclofosfamide, vincristina e prednisolone, oppure doxorubicina e fludarabina. Il trattamento dei linfomi non Hodgkin indolenti può provocare la remissione temporanea del linfoma, ma raramente conduce alla guarigione completa.
-
Linfomi aggressivi
Si impiegano chemioterapici, anticorpi monoclonali e corticosteroidi. In caso di recidiva il trattamento è la chemioterapia ad alte dosi, la radioterapia o il trapianto di midollo, che sono molto efficaci e riducono in breve tempo le dimensioni del linfoma.